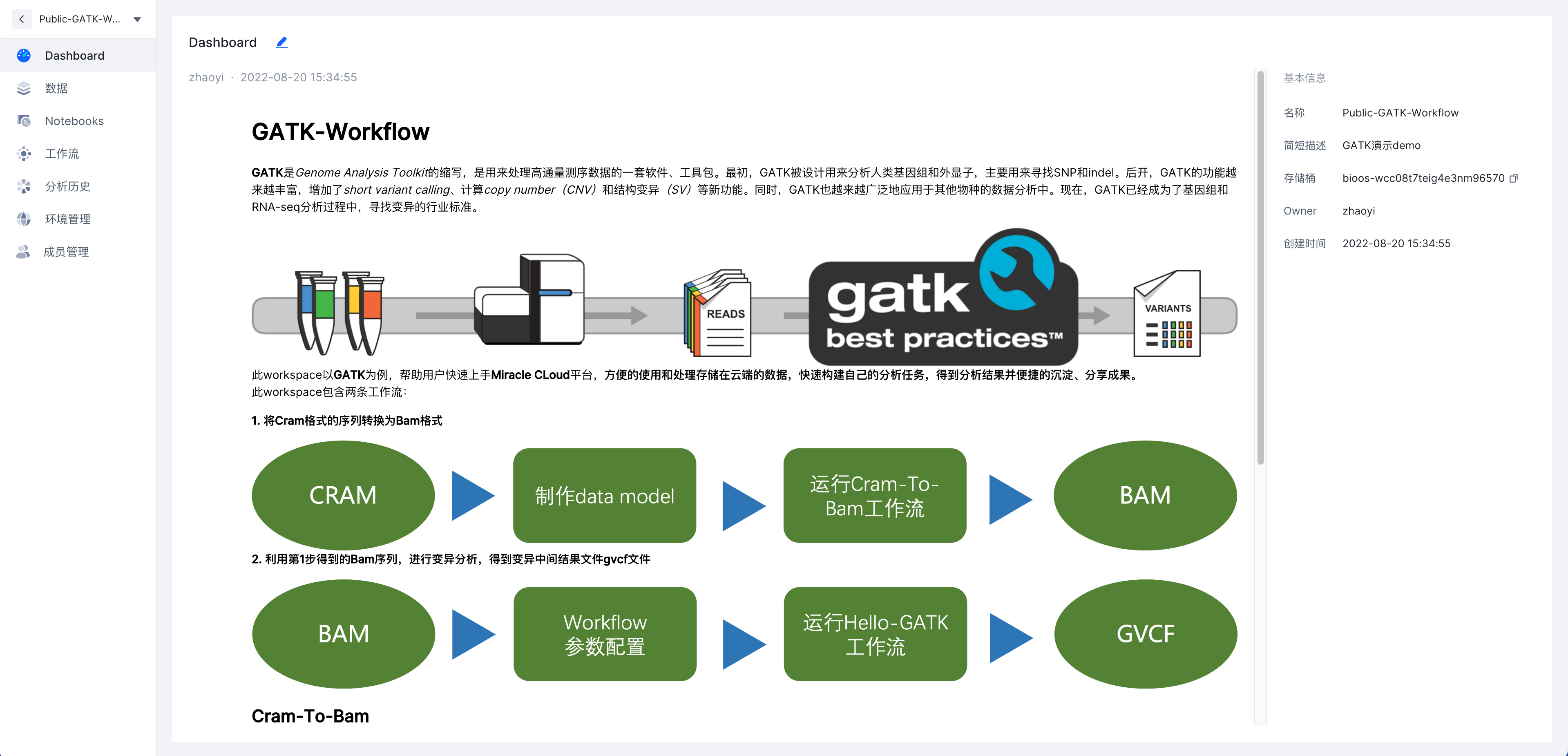
使用GATK进行基因组分析
本章节介绍了如何使用Genome Analysis Toolkit(GATK)在 Bio-OS 上运行基因组分析工作流。本章节中使用的工作流是 GATK ,用于将Cram格式的序列转换为Bam格式,并通过GATK,进行变异分析,得到变异中间结果文件gvcf文件。该工作流使用WDL编写,并通过 Cromwell 工作引擎调度运行。
GATK介绍
说明
GATK是Genome Analysis Toolkit的缩写,是用来处理高通量测序数据的一套软件、工具包。最初,GATK被设计用来分析人类基因组和外显子,主要用来寻找SNP和indel。后开,GATK的功能越来越丰富,增加了short variant calling、*Copy number variation(CNV)和结构变异(SV)*等新功能。同时,GATK也越来越广泛地应用于其他物种的数据分析中。现在,GATK已经成为了基因组和RNA-seq分析过程中,寻找变异的行业标准。
第一部分:运行预配置的数据格式转换(Cram2Bam)
您可以通过这部分了解数据的上传以及并成功运行工作流的方法。 该工作流程是一个文件格式转换,用于将基因组文件从一种格式 (CRAM) 转换为另一种格式 (BAM) 以进行下游分析。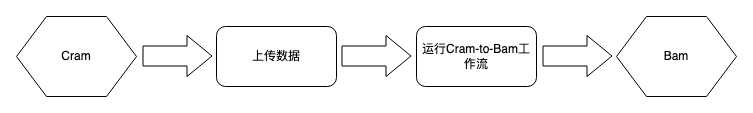
登录生信操作系统Bio-OS并创建共享集群
在左侧导航栏单击 【集群管理】
在 【共享集群】 标签页面,单击 【创建共享集群】
配置 workspace 环境
在左侧导航栏单击 【workspace】
在 【workspace 】 页面,单击 【新建workspace】
- 在弹出窗口页面,单击 【创建空白workspace】,名称为可以是GATK-workflow, 填写必要信息后选择 【确认】
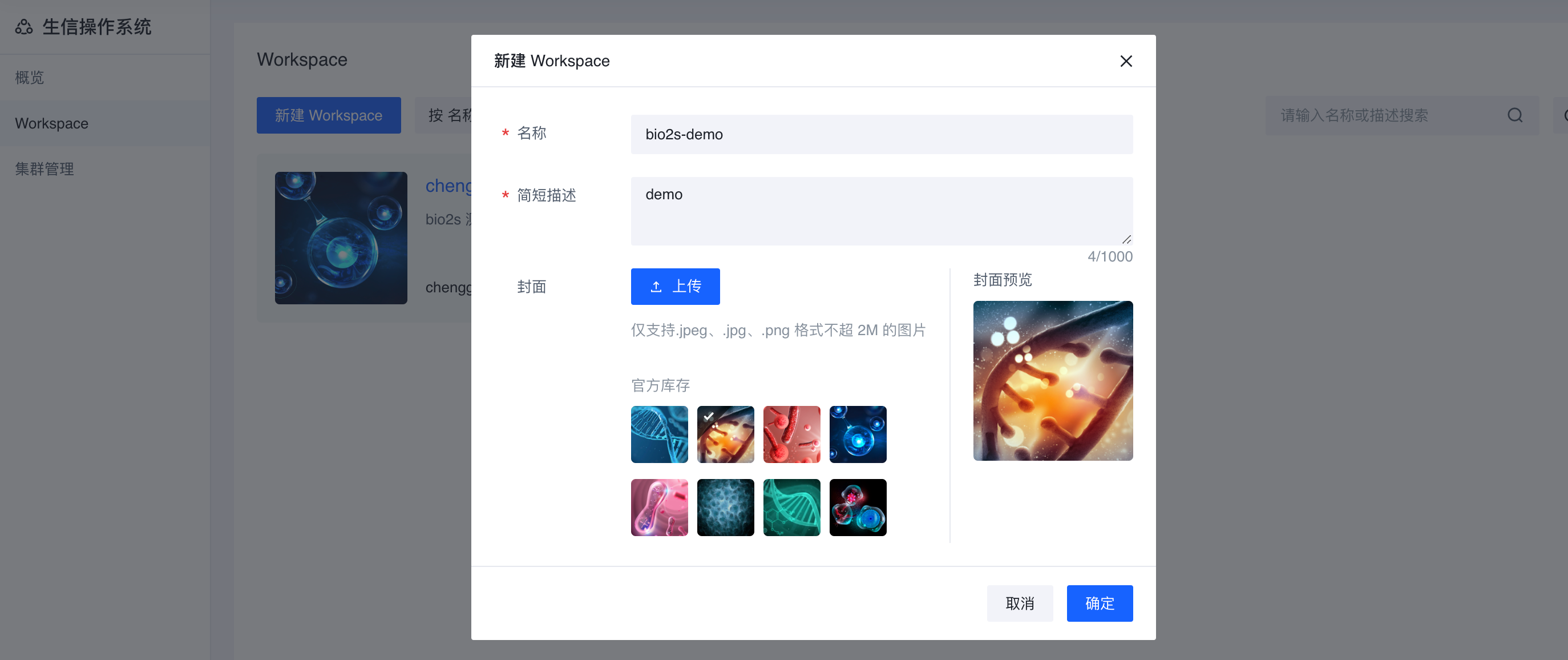
选择【我想留在当前页面】,点击刚刚创建的workspace ;
依次点击【环境管理】->【工作流】->【关联集群】,弹出页面中选择刚刚创建的集群;
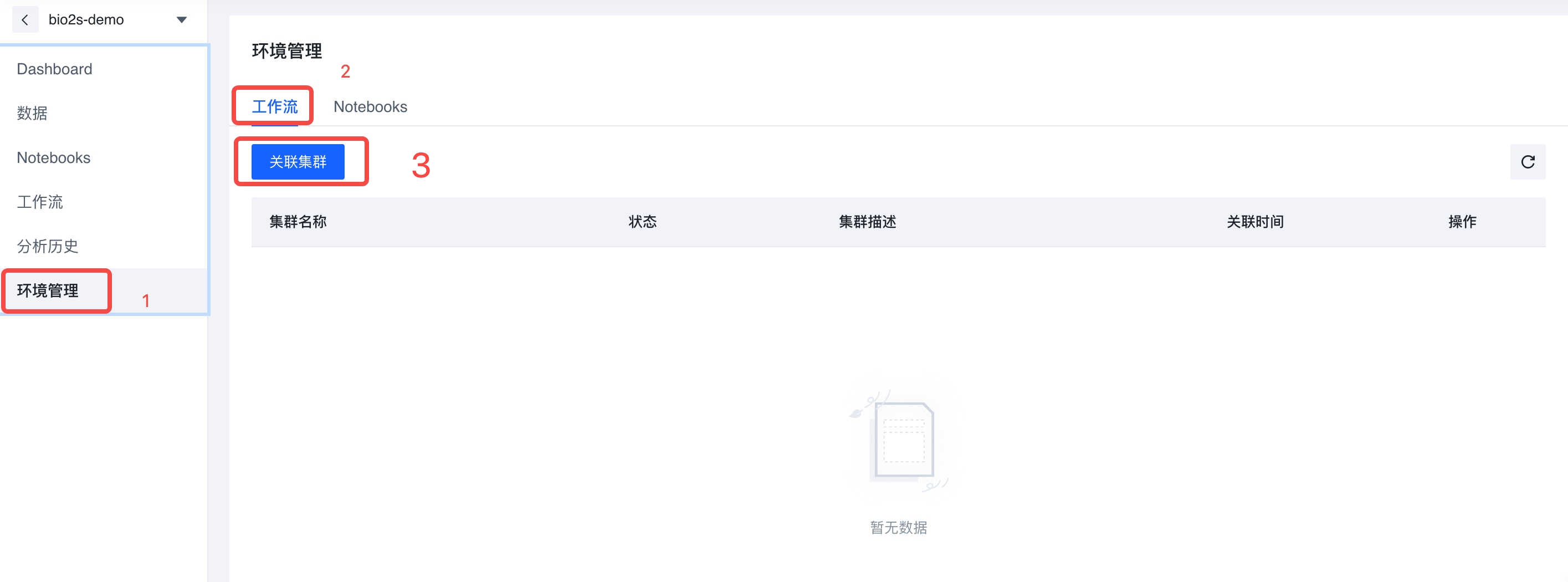
- 同理也对 【环境管理】->【notebooks】->【关联集群】执行相同操作。
上传数据
通常上传所需要分析的数据是开启一个工作流的第一步,在这个实践中,我们需要先将所需要用到的数据集传到TOS存储桶中(请确保您已经开通TOS对象存储服务),并手动生成一个CSV的数据模型表格。你需要将数据模型表格中所需关联的数据文件上传到Bio-OS的存储桶中。您有以下两种方式使用数据:
可以根据以下链接先下载样本数据集文件和参考数据集文件至本地,然后再上传至Workspace对应的存储桶中,最后可以根据文件对应的S3路径制作数据模型
可以直接下载此最佳实践对应的数据模型sample.csv文件,选用此方式可以直接从下面的步骤c开始
| 类型 | 数据地址 |
|---|---|
| 参考数据 | https://sample-data.tos-cn-beijing.volces.com/Homo_sapiens_assembly38.dict |
| https://sample-data.tos-cn-beijing.volces.com/Homo_sapiens_assembly38.fasta | |
| https://sample-data.tos-cn-beijing.volces.com/Homo_sapiens_assembly38.fasta.fai | |
| 样本数据 | https://sample-data.tos-cn-beijing.volces.com/NA12878.cram |
| https://sample-data.tos-cn-beijing.volces.com/my-sample-data.cram | |
| 数据模型CSV | https://sample-data.tos-cn-beijing.volces.com/sample.csv |
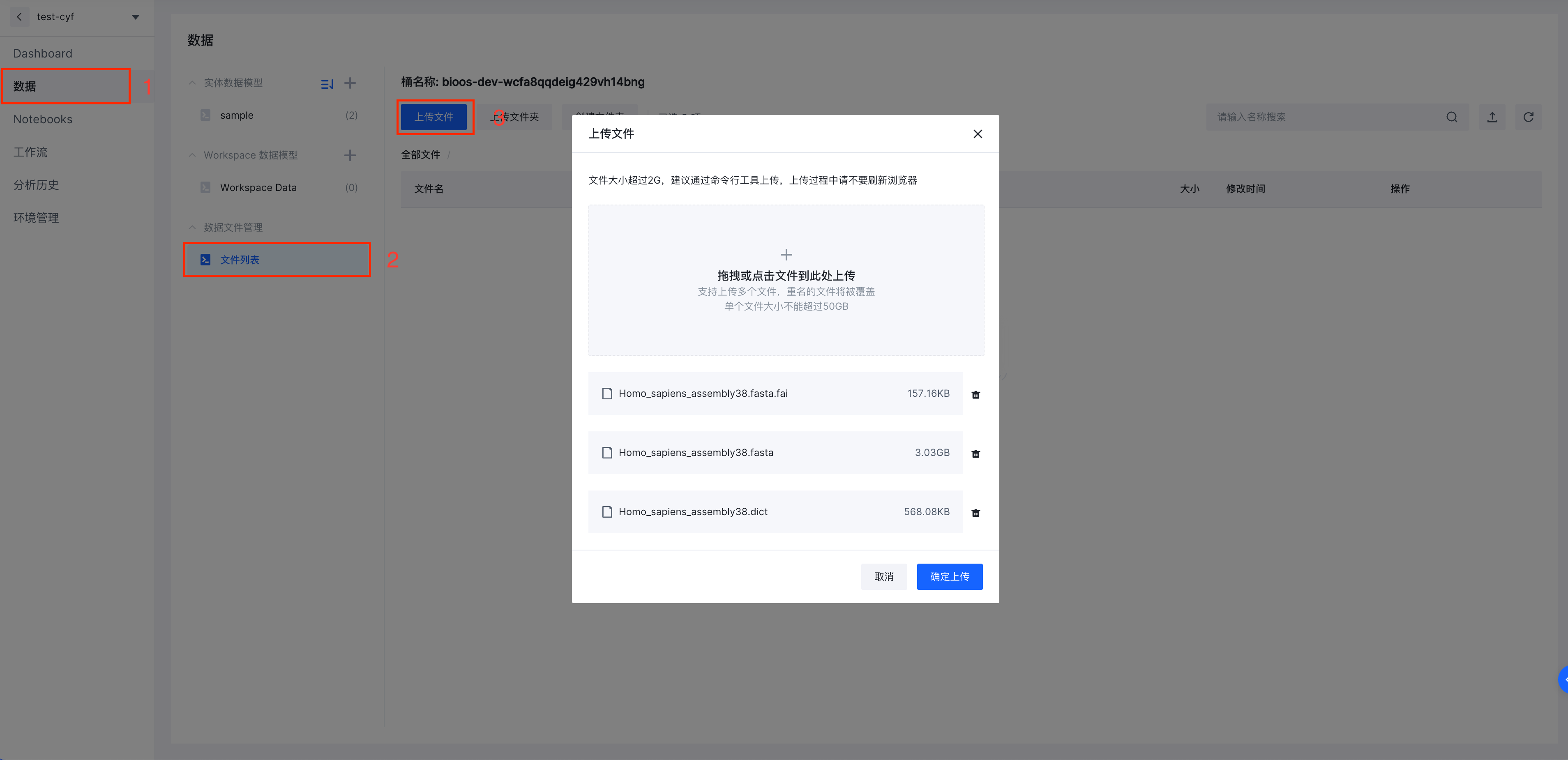
a. 点击平台左侧边栏数据-文件列表,进入文件数据管理页面,通过点击上传文件,可以根据引导上传本地数据文件至此Workspae对应的对象存储桶中(Workspace和对象存储桶为一对一关系)。
b. 数据上传完成后,下载csv模版并组织数据。点击左侧数据栏,点击 实体数据模型 列表右侧加号,出现上传数据模型选项框,点击下载数据模型模版。
c. 点击左侧数据栏,点击数据模型列表右侧加号,在这里我们使用下载csv模版并填写的方式进行数据组织。您可以直接将样本数据中的数据模型CSV进行上传。
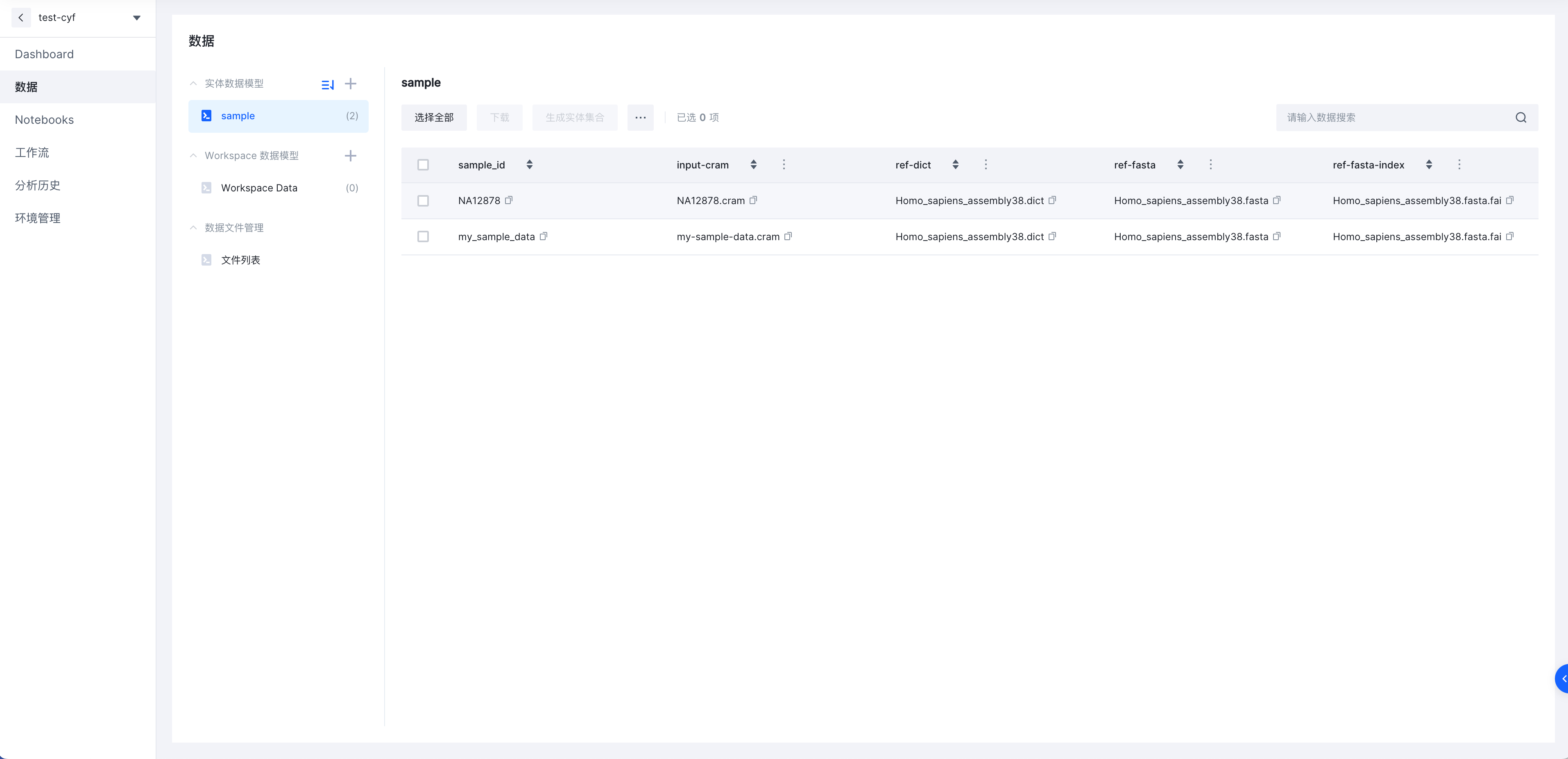
如下图可见,其中sample_id列为样本的唯一ID,之后的所有列皆为属性列,在属性列中,属性可以是具体的整数,字符串,当然更多的时候属性列会关联到云上的数据文件,在这里我们使用我们刚才存储在存储桶中的数据,并将数据链接在属性列中一一关联。

将编辑好的文件上传至Bio-OS,即可看到当前的上传的内容。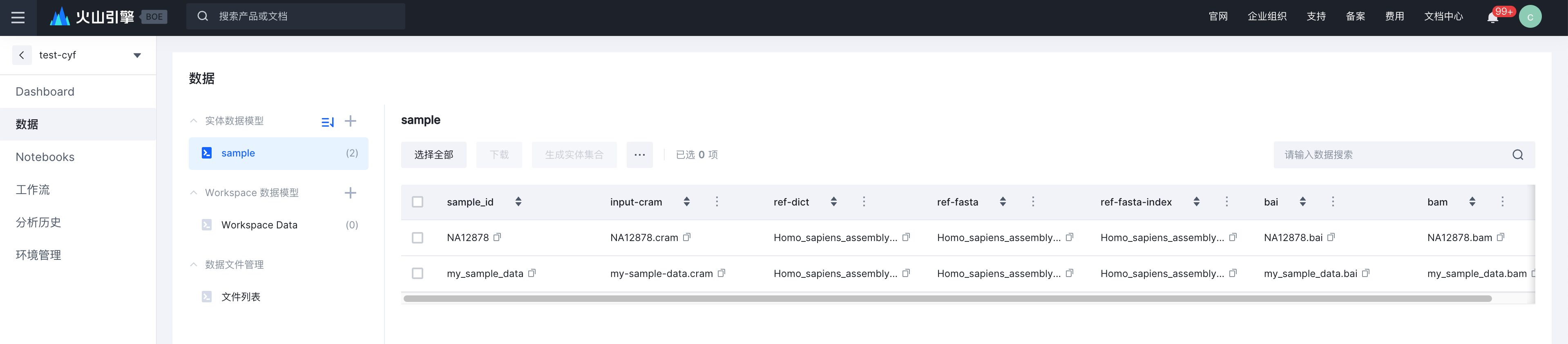
导入工作流
Bio-OS使用工作流描述语言 (WDL) 中的工作流来批处理基因组学数据。所有接收映射读取数据的 GATK 工具都期望 BAM 文件作为主要格式。有些支持CRAM格式,但我们观察到直接从 CRAM 文件工作时会出现性能问题,因此,我们首先将 CRAM 转换为 BAM。
说明
SAM、BAM 和 CRAM 都是原始 SAM 格式的不同形式,这些格式是为保存对齐(或更准确地说,映射)的高通量测序数据而定义的。SAM 代表序列比对图,并在此处的标准规范中进行了描述。BAM 和 CRAM 都是 SAM 的压缩形式;BAM(用于二进制对齐图)是一种无损压缩,而 CRAM 的范围可以从无损到有损,具体取决于您想要实现多少压缩(实际上最多)。BAM 和 CRAM 拥有与其 SAM 等价物相同的信息,结构方式相同;它们之间的不同之处在于文件本身的编码方式。
接下来我们需要将Cram-to-Bam 工作流导入到Workspace中,
点击导入工作流
选择自定义导入
输入对应的输入项(输入项的填写方法可查看用户指南中的*自定义导入*)
Git address: https://gitee.com/joy_lee/seq-format-conversion01
Tag: v0.47
Main path:CramToBam.wdl

这样就完成了我们所需要的Cram to Bam的转换工作流导入。
此处附WDL源码 Gitee 源码
version 1.0 #WORKFLOW DEFINITION workflow CramToBamFlow { input { File ref_fasta File ref_fasta_index File ref_dict File input_cram String sample_name String gotc_docker = "biocontainers/samtools:v1.7.0_cv4" } #converts CRAM to SAM to BAM and makes BAI call CramToBamTask { input: ref_fasta = ref_fasta, ref_fasta_index = ref_fasta_index, ref_dict = ref_dict, input_cram = input_cram, sample_name = sample_name, docker_image = gotc_docker } #Outputs Bam, Bai, and validation report to the FireCloud data model output { File outputBam = CramToBamTask.outputBam File outputBai = CramToBamTask.outputBai } } #Task Definitions task CramToBamTask{ input{ #Command parameters File ref_fasta File ref_fasta_index File ref_dict File input_cram String sample_name # Runtime parameters #Int addtional_disk_size = 20 Int machine_mem_size = 4 Int disk_size = 50 String docker_image } # Float output_bam_size = size(input_cram, "GB") / 0.60 # Float ref_size = size(ref_fasta, "GB") + size(ref_fasta_index, "GB") + size(ref_dict, "GB") # Int disk_size = ceil(size(input_cram, "GB") + output_bam_size + ref_size) + addtional_disk_size #Calls samtools view to do the conversion command { set -eo pipefail samtools view -h -T ~{ref_fasta} ~{input_cram} | samtools view -b -o ~{sample_name}.bam - samtools index -b ~{sample_name}.bam mv ~{sample_name}.bam.bai ~{sample_name}.bai } runtime { docker: docker_image memory: machine_mem_size + " GB" disk: disk_size + " GB" } #Outputs a BAM and BAI with the same sample name output { File outputBam = "~{sample_name}.bam" File outputBai = "~{sample_name}.bai" } }
运行工作流
刚才我们已经完成了工作流的导入,那么接下来我们将对导入的工作流进行运行。
选择我们刚才导入的Cram-to-Bam工作流
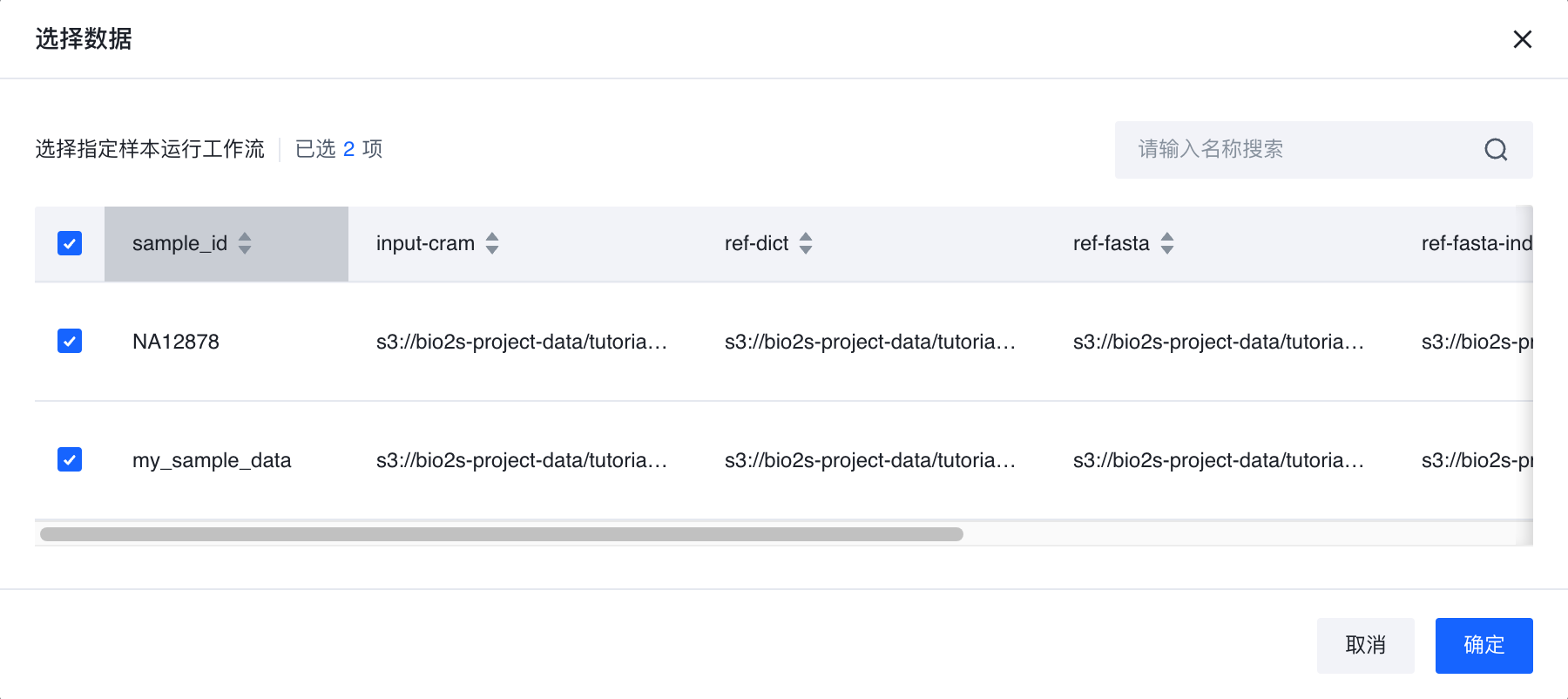
在这里我们需要配置运行选项和运行参数,在运行选项中,选择刚才我们第一步中所上传的数据实体,并指定实体数据。
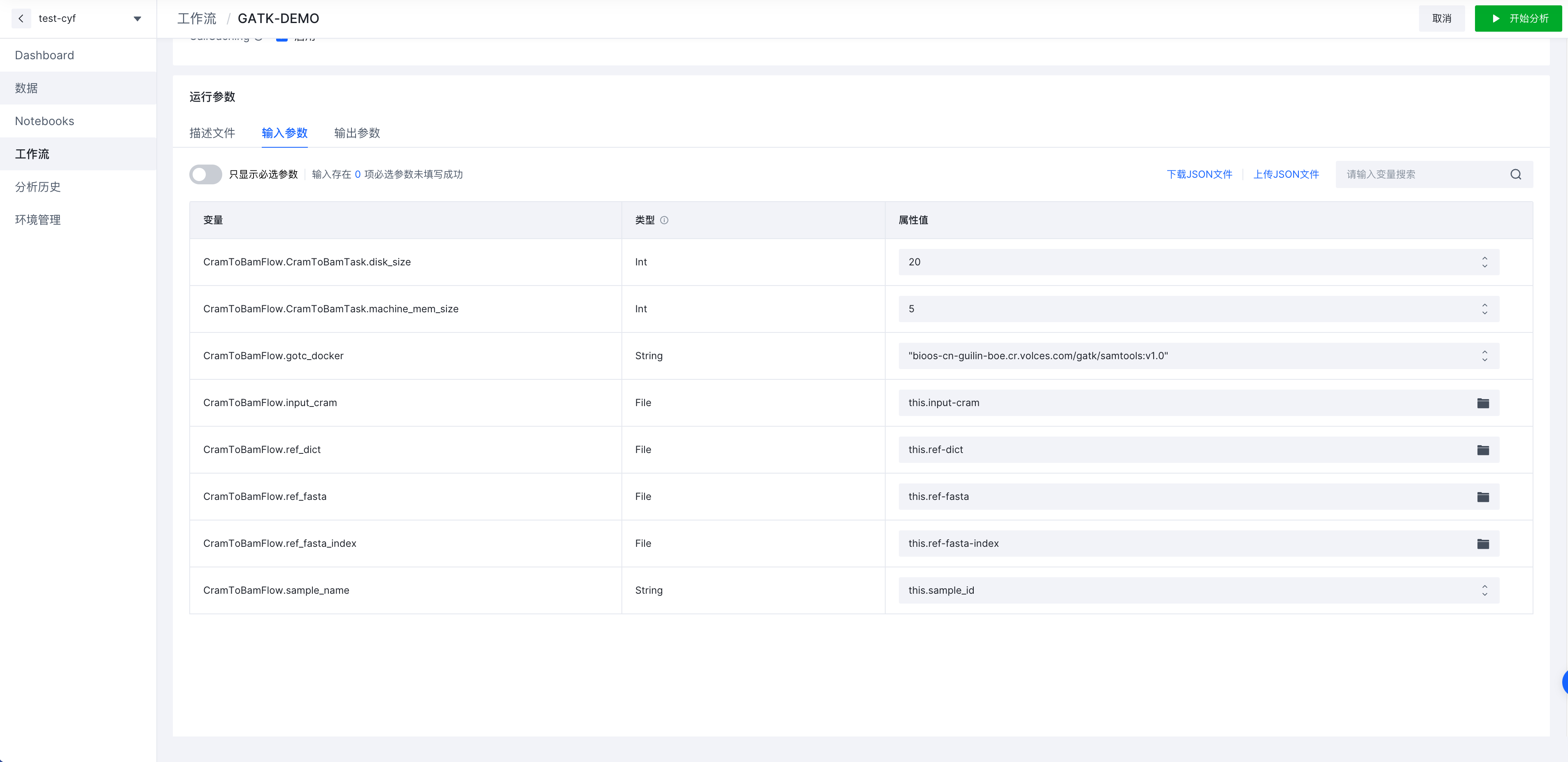
- 配置输入参数,选中输入参数选项卡,并按照如下参数进行输入,当然当输入参数较多时,可以使用上传JSON文件的方式进行快速地导入。
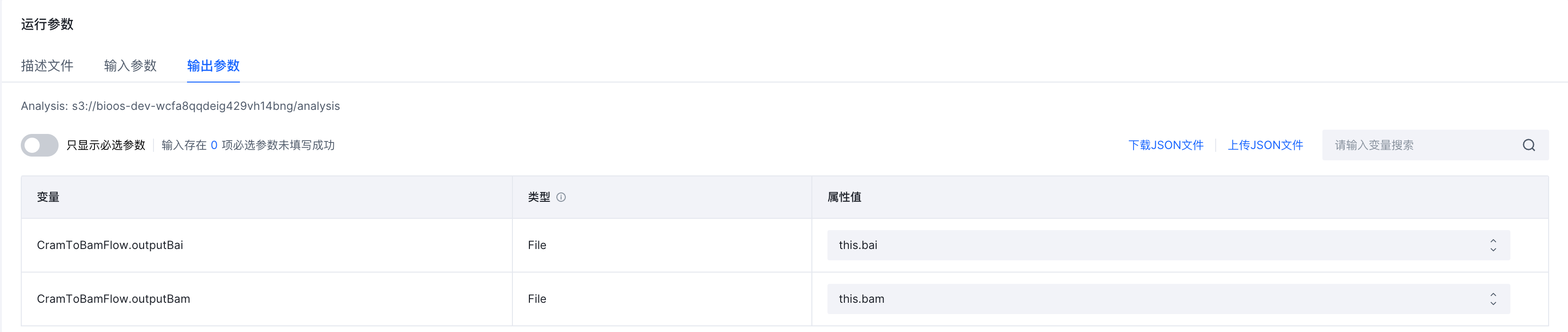
- 配置输出参数,使用this.bai作为bai文件的输出属性,this.bam作为bam文件的属性列
通过this.列名,定义输出的结果(TOS链接或数组)输出到所选择的数据表的指定列中;如果原表格中没有此列则按照列名新加列;如果原表格有对应的列,则会将直接将新结果进行填充或覆盖
- 点击右上角【开始分析】,此时工作流会通过Cromwell工作流引擎进行分析,结果可在【分析历史】中进行查看
第二部分:对生成BAM输出通过GATK进行进一步分析
这个章节中我们需要使用第一部分所输出的Bam以及Bai文件通过GATK工作流进行进一步分析,最终输出所需要的gvcf文件。
导入GATK工作流
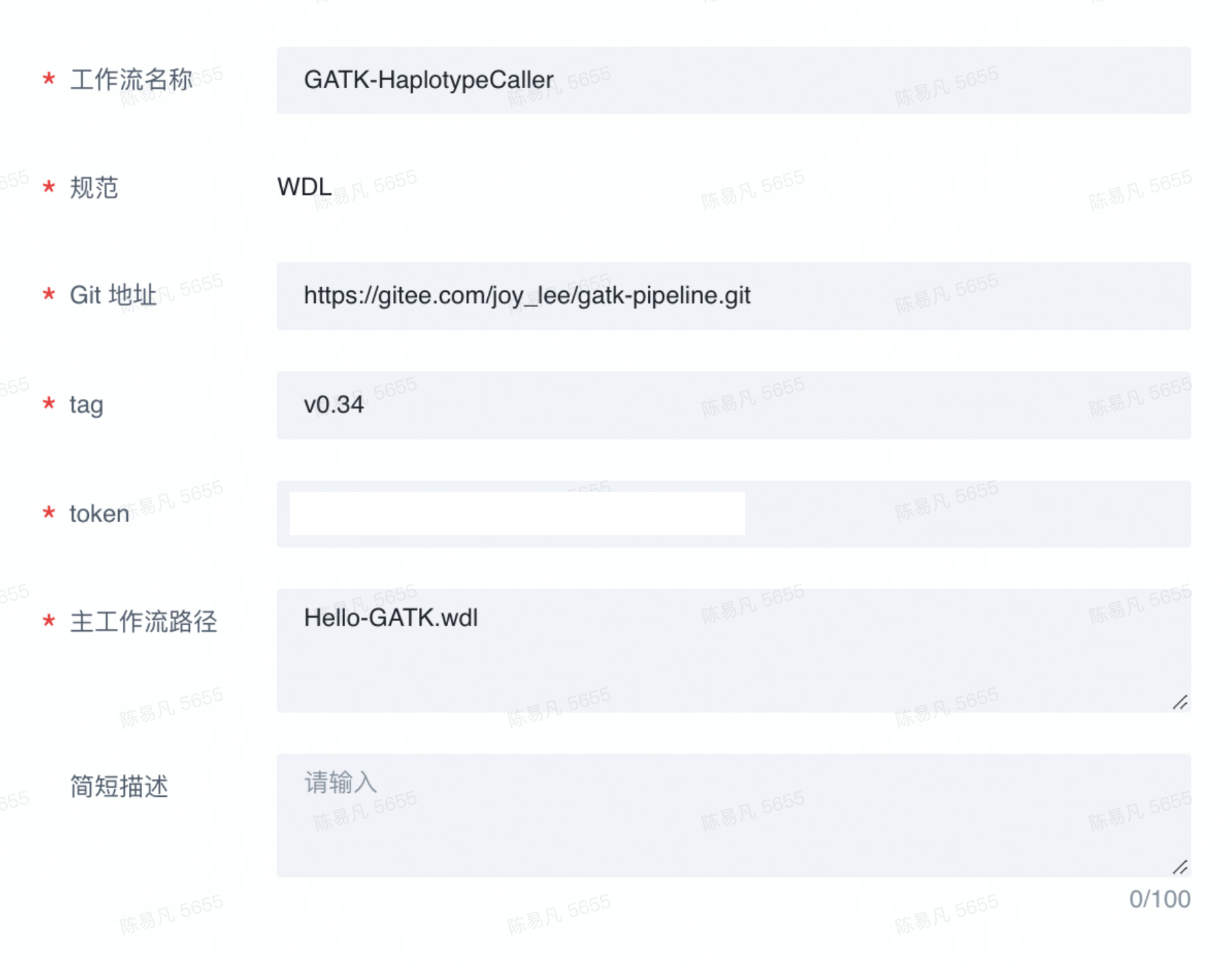
我们还需要导入针对Bam序列进行进行变异分析,得到变异中间结果文件gvcf文件的工作流。点击左侧【工作流】->【导入工作流】, 填写对应的输入信息:
Git address:https://gitee.com/joy_lee/gatk-pipeline
Tag: v0.34
Main path:Hello-GATK.wdl
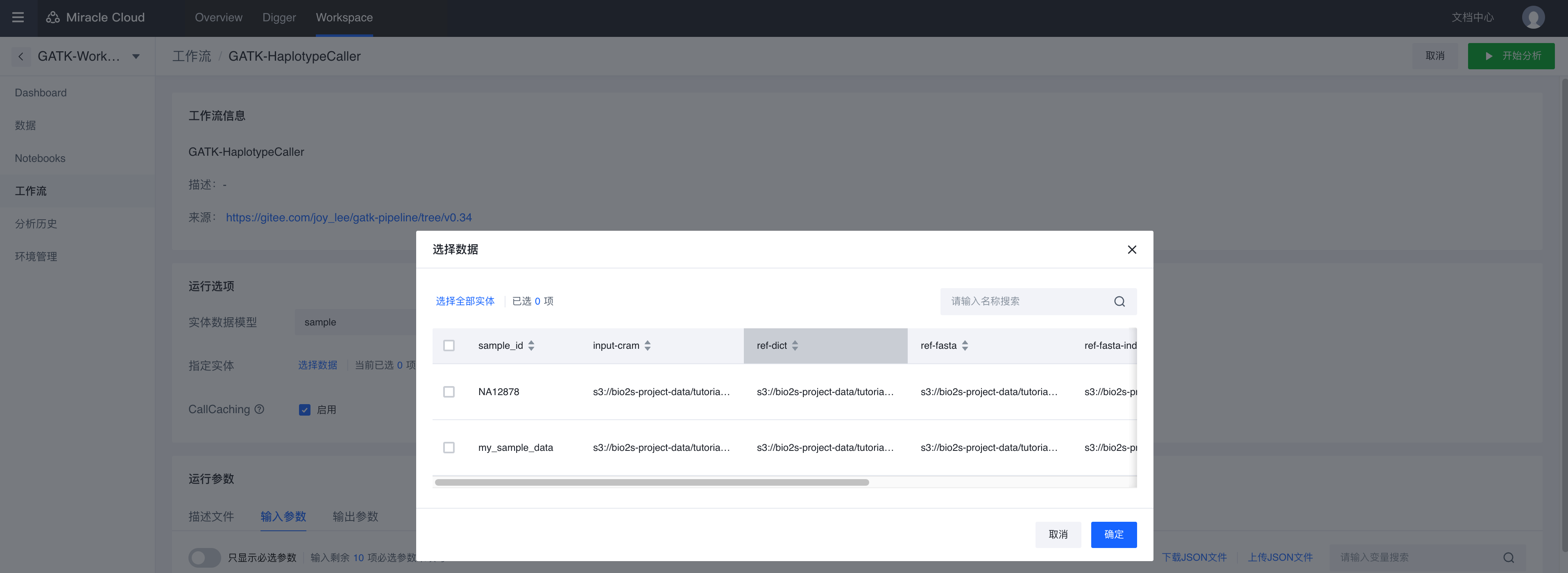
运行GATK工作流
- 选择数据: sample,仍然选择实体样本NA12878和my_sample_data,这部分我们将使用第一部分中所输出的bam文件作为这一部分的输入。
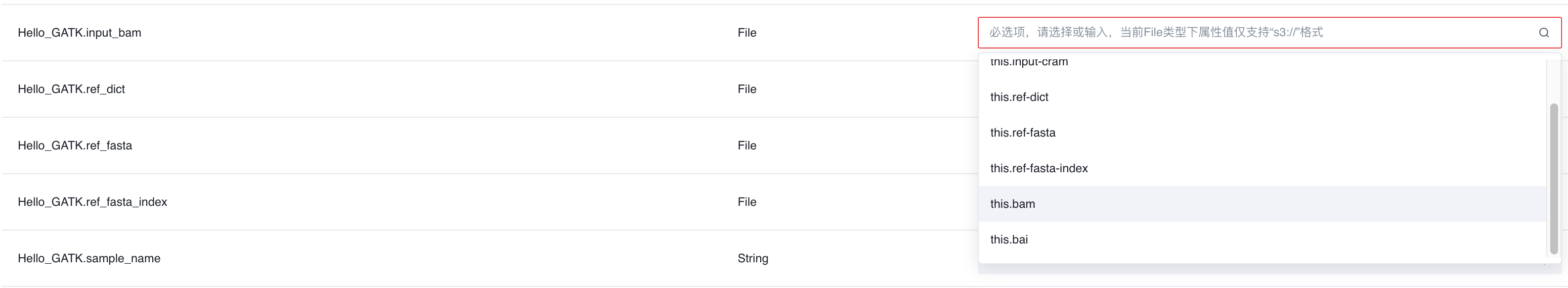
- 点击 【输入参数】 选项卡 ,选择Hello_GATK.input_bam, 在属性值下拉选择this.bam
将所有输入配置完成后如下所示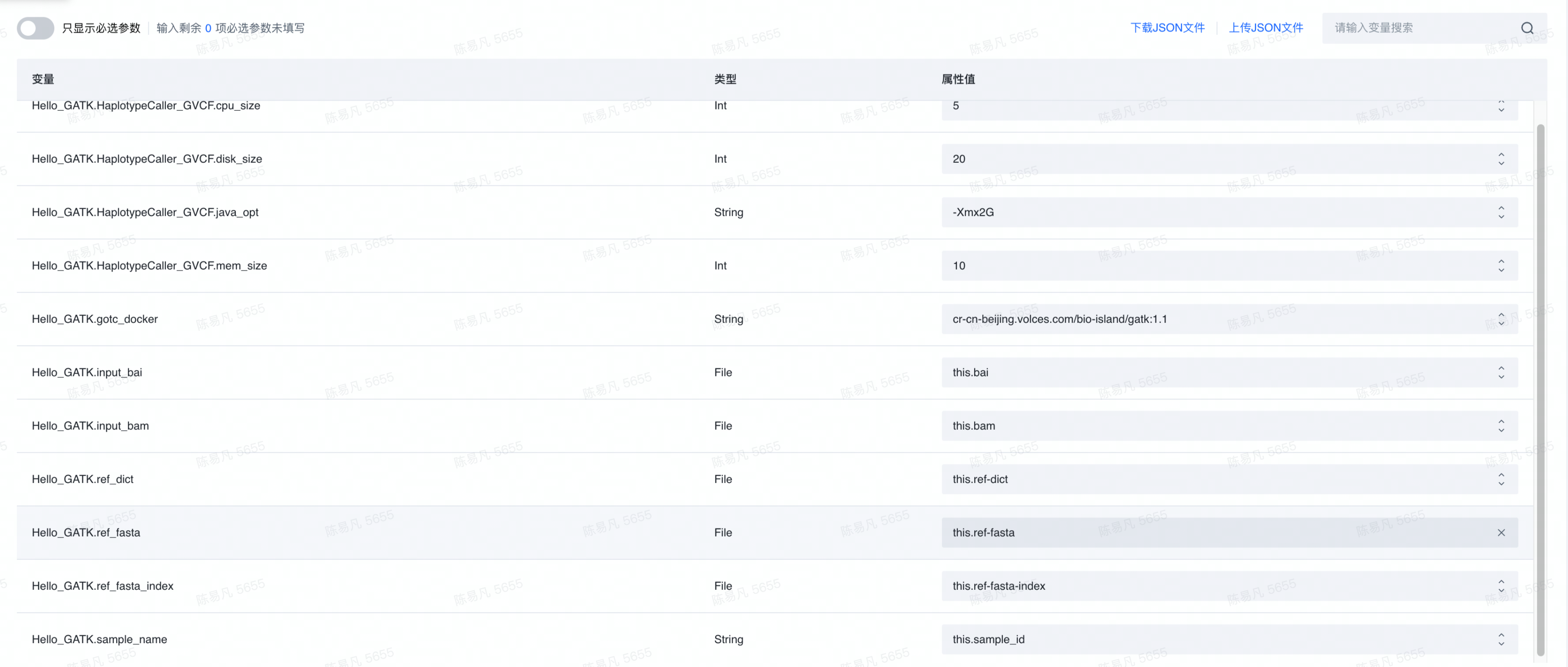
- 点击 【输出参数】 选项卡,您可以在其中填写 output_gvcf 变量的属性。要写入数据表,首先键入“this.” ,然后为该属性添加一个名称。工作流将在sample数据表中生成新的一列。
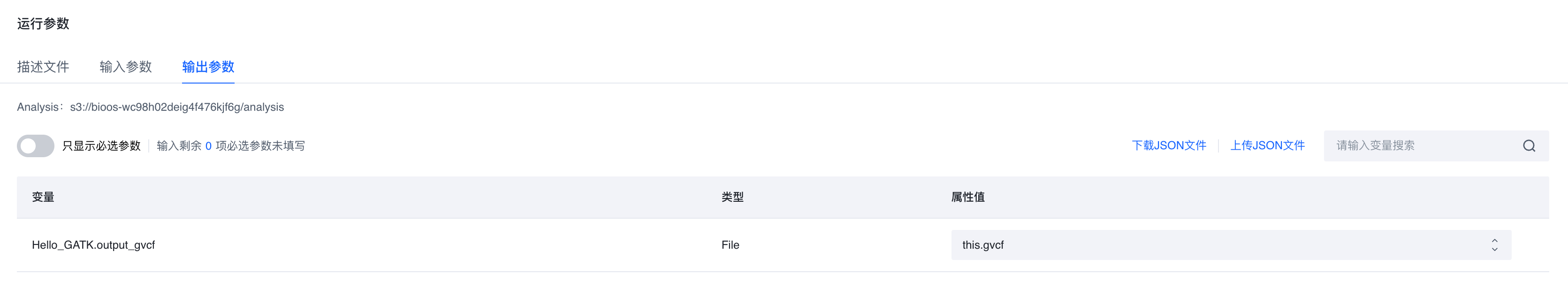
- 点击右上角 【开始分析】 按钮以提交您的工作流
如果您的结果正确提交,那么在分析历史中查看到当前工作流的分析状态。稍等一段时间之后您就会看到分析状态转为分析成功
查看分析历史
等待分析完成之后可以在点击左侧菜单栏【分析历史】,点击选择刚完成的分析投递,点击右侧【查看】输出参数,并将对应样本的gvcf文件进行下载。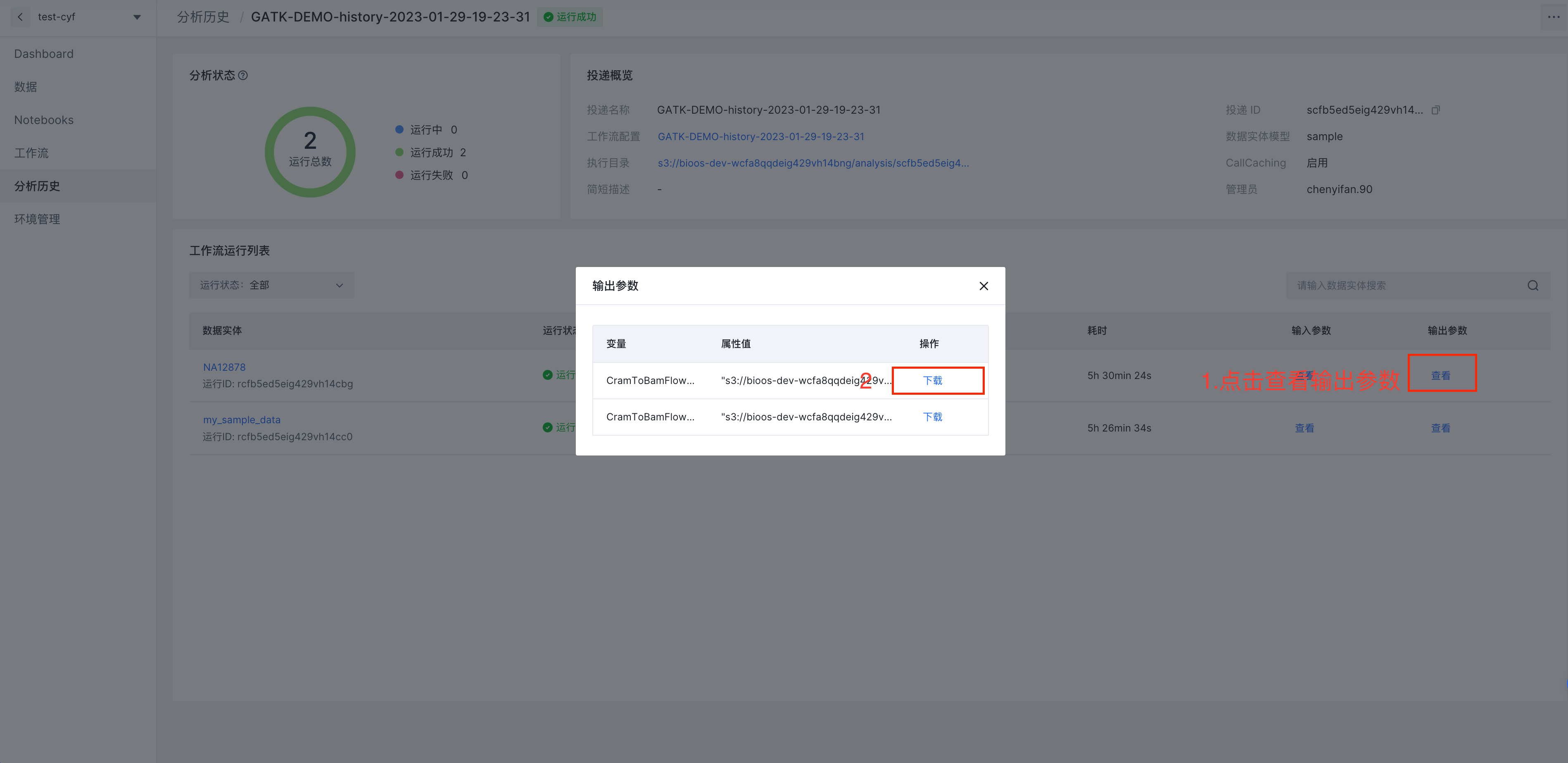
至此我们也完成了两个工作流的运行,完成了从Cram到Bam文件的格式转换,并通过GATK对Bam序列,进行变异分析,得到变异中间结果文件gvcf文件。
第三部分:完成Workspace概述
那么至此如果您已经完成了所有的工作流和分析工作,您可能还希望为您的Workspace写一个摘要或者介绍,您可以点击左侧Dashboard,Dashboard同样集成了Jupyter Notebook,您可以使用Jupyter Notebook中使用MarkDown进行文档的编写。(Markdown文档的编辑方法可参看:如何在Markdown单元格中编辑内容)
完成编辑后点击保存,结果如所示: